SRAデータを活用した再解析、スプライシング異常の一つ「スプライスサイト生成変異」に着目
国立がん研究センターは1月9日、難病・がん領域で非常に重要な変異のタイプである「スプライスサイト生成変異」を、トランスクリプトームシークエンスデータから同定する新規情報解析手法(juncmut)を開発したと発表した。この研究は、同センター研究所ゲノム解析基盤開発分野の白石友一分野長らの研究グループによるもの。研究成果は、「Nature Communications」にオンライン掲載されている。
画像はリリースより
(詳細は▼関連リンクからご確認ください)
ハイスループットシークエンス技術の革新、またその有効性が広く認められたことで、オミクスデータの蓄積が国際的に加速している。これらのデータを集積したレポジトリを活用し、世界中の研究者が新たな視点でシークエンスデータの再解析を行うことで、データの潜在的な価値を最大限に引き出そうとする取り組みが世界中で進められている。その中でも特に重要なデータレポジトリの一つが「Sequence Read Archive(SRA)」である。SRAは、主に次世代シークエンサーにより生成されたゲノム、トランスクリプトームなどの膨大な配列データを蓄積したもので、公開されている多くのデータは誰でも自由に利用可能である。18か月ごとに収録データが倍増しており、今後もさらなる拡大が見込まれている。
研究グループはこれまでに、SRAに登録されたトランスクリプトームシーケンスデータを活用して、スプライシング異常を介した疾患関連のゲノム変異を効率的に同定するためのプラットフォームを開発してきた。過去の研究では、イントロン残存というスプライシング異常を引き起こすゲノム変異に注目した。一方で、今回の研究では、スプライシング関連変異の中でも特に重要とされる「スプライスサイト生成変異」に焦点を当てている。スプライスサイト生成変異はゲノム変異により新たなスプライスサイトが形成されることで、異常なスプライシングパターンを引き起こす。
スプライスサイト生成変異を同定することは非常に難しく、疾患ゲノム解析の中で多くの場合見逃されてきた。しかし、これらの変異をデータベース化することで、これまで解明されていなかった疾患の原因となる遺伝子変異を発見できる可能性が広がる。また、スプライスサイト生成変異は、核酸医薬を用いた治療の有望なターゲットとして注目されている。このデータベースを構築することで、希少疾患を抱える患者に対する個別化治療の進展が期待される。
トランスクリプトームデータのみでスプライスサイト生成変異を同定するアルゴリズム開発
スプライシング異常を引き起こすゲノム変異の同定には、ゲノムデータとトランスクリプトームデータの両方が必要とされてきた。しかし、このようなペアデータが存在しているサンプルは非常に限られている。そこで研究グループは、SRAが提供する膨大なトランスクリプトームシークエンスデータを最大限に活用するため、ゲノムデータなしでトランスクリプトームシークエンスデータのみを用いてスプライスサイト生成変異を同定するアルゴリズム「juncmut」を開発した。
32万件のデータから、3万件のスプライスサイト生成変異を同定しカタログ化
研究グループは、国立遺伝学研究所のスーパーコンピューターを用い、32万2,072件のトランスクリプトームシークエンスデータをjuncmutで再解析した。その結果、合計3万130件のスプライスサイト生成変異を同定し、それらをカタログ化することに成功した。収集した変異リストは新たに開発されたポータルサイト、SSCV DB(https://sscvdb.io)に公開されている。
Aluエキソン化を詳細に解析、特に偽エキソン形成されやすいAlu配列内領域が判明
この大規模な変異カタログを活用し、研究グループはスプライスサイト生成変異に関するさまざまな性質や現象を調査した。その一例として、「Aluエキソン化」に関する特性を詳細に解析した。Alu配列は約300bpの配列で、元来エキソンを形成しやすい性質を有している。イントロン領域に挿入されたAlu配列は、さらにスプライスサイト生成変異を獲得することにより、新しいエキソンを形成する。この現象は「Aluエキソン化」と呼ばれ、霊長類のゲノム進化の文脈で研究されてきた。近年では、希少疾患やがんの進展においても、同様の現象が生じていることが明らかになってきた。研究グループは、Alu配列上にある3,102件のスプライスサイト生成変異を調査し、「偽エキソン」が特に形成されやすい領域を特定した。特に最も「偽エキソン」が形成されやすいのは、Alu配列の157番目から始まり280番目で終わる領域だった。
がん関連NOTCH1、変異による異常スプライスサイト標的の核酸医薬設計し効果を実証
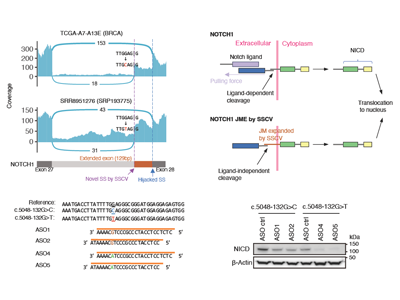
また、得られたスプライスサイト生成変異の中で、がんの進展に関与すると考えられるNOTCH1遺伝子の変異に注目した。この変異は、NOTCH1遺伝子のexon 28から132bp上流に位置するゲノム変異であり、これによってトランスクリプトが129bp延長され、juxtamembrane領域に43アミノ酸の挿入を引き起こす。この変異により、NOTCH1がリガンド非依存的に活性化されることが予想された。研究グループは、この変異を再現するCRIPSRゲノム編集細胞モデルを開発し、NOTCH1の顕著な活性化を確認した。さらに、この変異により形成されるスプライスサイトを標的とする核酸医薬を設計して投与することで、この異常な活性化を効果的に抑制できることを実証した。
新規疾患関連・創薬標的の変異を自律的に同定する仕組みの構築につながると期待
今回の研究では疾患に関連している遺伝子上に計5,121個のスプライスサイト生成変異が同定されており、これらのほとんどはこれまでに検出されていない、または疾患に関連が知られていなかった変異である。この中には、今回の研究で詳細に検証を加えたNOTCH1遺伝子の変異の他にも、さらに多くの疾患に関連した変異、創薬標的となる変異が含まれていることが予想される。
また、SRAなどのデータベースにおいては、現在進行形で新たなデータの蓄積が進んでいる。今後加速度的に蓄積が進むトランスクリプトームデータに対して、今回の研究で開発した情報解析プラットフォームを適用することで、さらに多くの新規疾患関連・創薬の標的となる変異を自律的に同定する仕組みを構築することが期待できる。
今回の研究によりスプライスサイト生成変異は多数検出できるようになったが、その一つ一つが実際に疾患に関連しているか、創薬のターゲットになるかについては、専門家が個別に情報を検証し解釈をする必要がある。「今後、生成AIを活用し、アノテーションシステムの精緻化を図ることで、疾患や創薬に重要な変異を効率的に特定するアプローチの開発を計画している」と、研究グループは述べている。
▼関連リンク
・国立がん研究センター プレスリリース

